Last week, I took a lighter turn on things, talking about the lighter side of ISO. Some of the more interesting ISO standards out there, like shoe size, traffic lights, and snorkeling etiquette. I also posted a poll on the “state of the standards”, and here are the results.
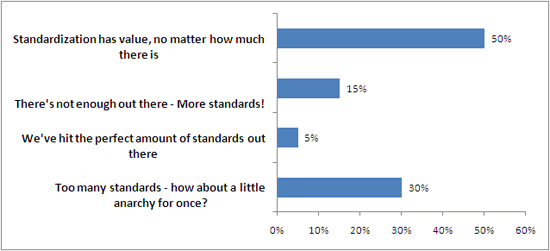
You cheeky anarchists, you.
All kidding aside, one of the things that make ISO quality standards so useful is their correlation amongst each other. Many ISO standards, especially those that deal with management, are based off each other. This can come in handy when looking to add new management systems to your organization. Chances if you are already ISO 9000 compliant, you have most of the requirements covered in other management standards.
I’ve written in the past about the Convergences of QMS and EHS, and one of the things that always comes up is the daunting task of figuring out where the correspondence is. While not every organization is on the ISO standardization train, we can look at the three ISO standards that govern QEHS and see how well they fit together. The three Standards i speak of are:
ISO 9001: Quality Management Systems – This standard involves people-oriented processes as well as customer requirements, and ensures that all services or products adhere to customer requirements.
ISO 14001:Environmental Management Systems – This standard helps analyze an organization’s activities to determine which areas the environmental impacts can be improved. In addition, requirements are driven by the organization implementing the standard, as opposed to customer-focused requirements
OHSAS 18001: Health and Safety Management Systems – This standard has direct similarities to the ISO 14001 standard, but outlines the processes for Health and Safety Management. An EHS system combines the ISO 14001 and OHSAS 18001 standards into one solution.
(yes the third is not technically an “ISO” standard, but is widely based on the ISO model, as you’ll see in a moment)
Many organizations that use ISO 9000 systems are now choosing to also implement ISO 14001 and OHSAS 18001 compliant systems due to factors such as demands of government regulations, customers, risk management, market requirements and corporate commitment. Recent reports from analyst firms such as Gartner Research have confirmed that the market is experiencing this convergence.
Although these ISO and OHSAS standards were developed to serve different purposes, they share many similarities. In fact, ISO 9000 was one of the source documents used for ISO 14001, making the two standards very compatible. OHSAS 18001 and ISO 14001 are essentially identical, making implementation of a converged system less complex. Both the ISO 14001 and the OHSAS 18001 standards are based on the Plan, Do, Check, Act (PDCA) cycle. While ISO 9000 also follows PDCA, it is more process focused.
When taking a high-level look at these three standards, it is clear that there is significant overlap. An organization can map the requirements across these three standards into a single, consolidated requirements set. Take a look at this graphic on how the three standards overlap:
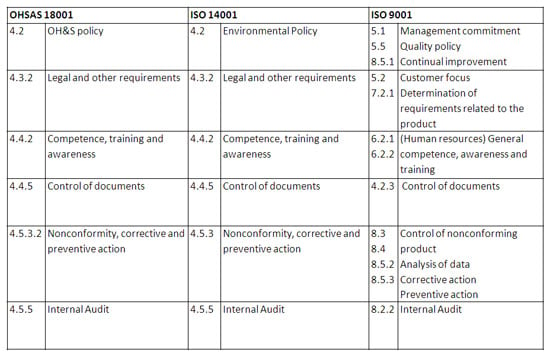
Best in class solutions recognize this overlap in functions, and are able to create a single, holistic system that not only provides depth in the specialized functions, but also is equipped with a wide breadth of cross-functional functions that serve the needs of both Quality and EHS.
Some of these cross-functional integrated processes include:
• Audits and Surveys: The cross-functional Audits and Surveys function enhances efficiency by automating the process of auditing and customer satisfaction surveys in both QMS and EHS systems. The Audits and Surveys function has additional features including a question library and ready-to-use checklists for both ISO 9000 and ISO 14001 standards. If needed, CAPA is initiated as a result of audit findings and audit information will automatically be inherited into the CAPA form.
• Corrective and Preventive Action: In any organization it is essential to effectively track the cost and cause of any discrepancies to mitigate the risk of recurrence. The CAPA function can be utilized by both EHS and QMS systems to efficiently track these discrepancies. Relevant data is automatically inherited for each system—for EHS this includes Incidents and Audits and for QMS it includes Nonconforming Materials (NCMR) and Calibration and Maintenance. Like all modules in an automated QEHS, CAPA integrates with additional modules in the product, including Risk Assessment, a key function in the CAPA process.
• Risk Assessment: In both QMS and EHS-centric organizations, efficiently managing risk ranks high in priority. The cross-discipline Risk Assessment function calculates risk at various points in a process and then displays this risk mitigation history by event. It integrates with the QMS or EHS CAPA function to filter the non-critical from the critical events and to ensure that the high-priority CAPAs are corrected first.
• Document Control: This function manages the lifecycle of all documents, which is beneficial for both QMS and EHS. Its centralized repository is an important aspect of this function as it holds all documents and electronic distribution of all released documents. The Document Control software integrates with other QMS functions such as Complaint Handling and Material Returns, and EHS functions such as Material Safety Data Sheet (MSDS) and Emergency Preparedness. This ensures proper documentation within the different processes of each system.
• Training: Employee Training solutions provide a simple way to schedule and record training for EHS and QMS-centric organizations. It tracks employee profiles, schedules training events and manages the identification, responsibilities, authorities, training and certification requirements for each employee.
There are plenty more areas of convergence in standards – these are just a few. Standards are a governing body for making management systems and manufacturing processes in-sync – like a big orchestra. By looking at the various standards and identifying the correspondence, you can create a holistic, converged management system.




